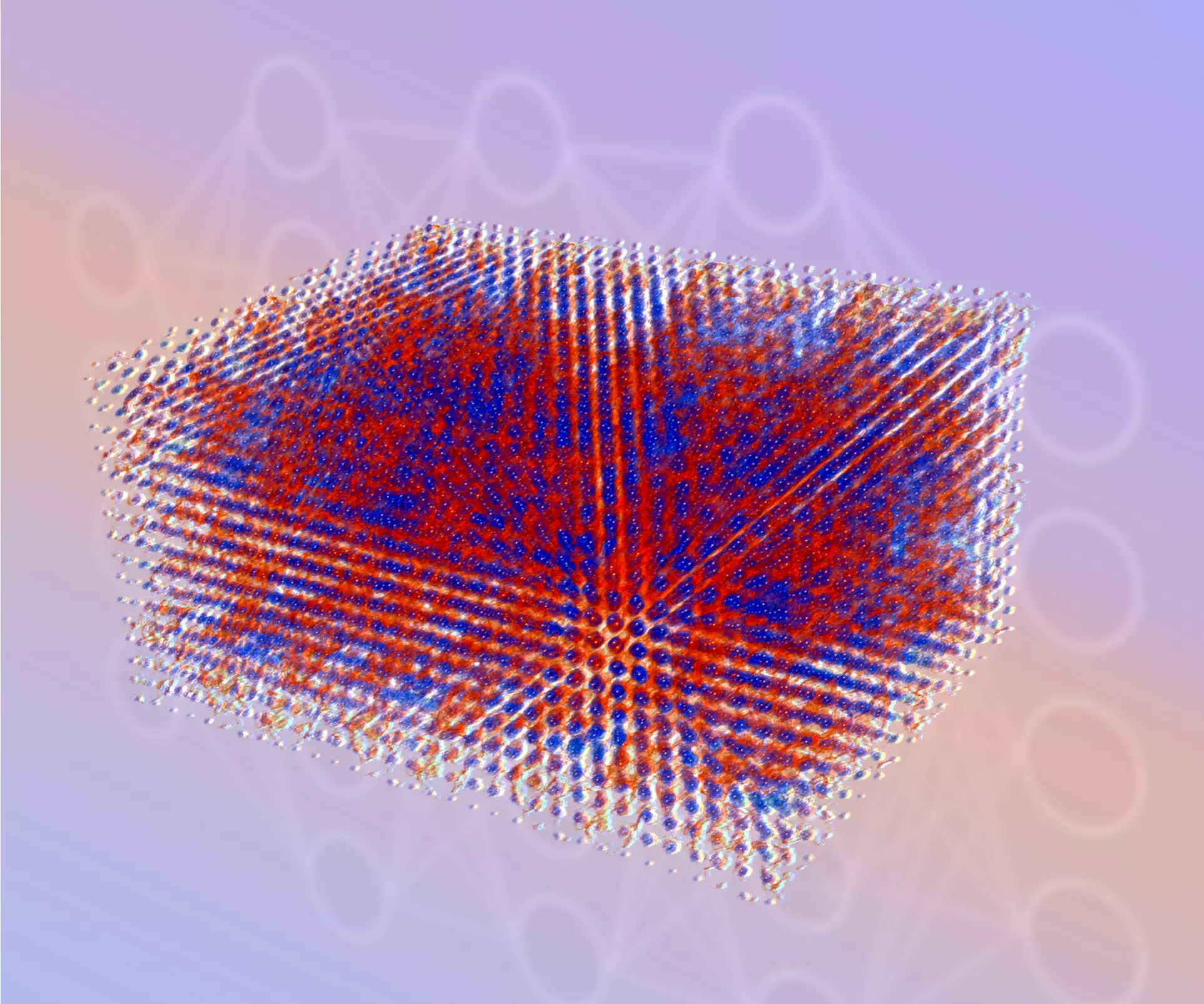
The electronic structure, which refers to the arrangement of electrons in matter, is a critical factor in both fundamental and applied research. From drug design to energy storage, understanding the behavior of electrons is essential. However, the lack of a simulation technique that can provide both accuracy and scalability across different scales has hindered progress in these fields. To overcome this challenge, researchers from the Center for Advanced Systems Understanding (CASUS) at the Helmholtz-Zentrum Dresden-Rossendorf (HZDR) in Germany and Sandia National Laboratories in the United States have developed a machine learning-based simulation method called Materials Learning Algorithms (MALA). This innovative approach surpasses traditional electronic structure simulation techniques and opens up new possibilities for scientific advancements.
Overcoming Limitations of Existing Methods
Previous simulation methods have faced limitations in accurately representing the quantum mechanical interactions of electrons. Classical atomistic simulation methods can handle complex systems, but they do not consider quantum electronic structure, limiting their applicability. On the other hand, first principles methods, such as density functional theory (DFT), provide high fidelity but are computationally demanding and restricted to small scales. The MALA software stack addresses these limitations by integrating machine learning with physics-based approaches. It combines deep learning, a machine learning method, with physics algorithms to predict the electronic structure of materials accurately.
Advantages and Achievements of MALA
The MALA software stack takes the arrangement of atoms in space as input and generates bispectrum components, which encode the spatial arrangement of atoms. The machine learning model in MALA is trained to predict the electronic structure based on these components. One significant advantage of MALA is that its machine learning model is independent of system size, allowing it to be trained on small systems and deployed at any scale. The researchers demonstrated the effectiveness of MALA by achieving a speedup of over 1,000 times for smaller system sizes compared to conventional algorithms. They also showcased MALA’s capability to accurately perform electronic structure calculations at a large scale, involving more than 100,000 atoms. This accomplishment was achieved with modest computational effort, highlighting the limitations of conventional DFT codes.
Attila Cangi, the Acting Department Head of Matter under Extreme Conditions at CASUS, believes that MALA will revolutionize electronic structure calculations. With MALA, researchers can simulate significantly larger systems at an unprecedented speed, allowing them to address a wide range of societal challenges. This includes developing new vaccines, studying material defects, exploring chemical reactions, and more. Additionally, MALA’s approach is highly compatible with high-performance computing (HPC). As the system size increases, MALA can leverage HPC resources, particularly graphical processing units, for efficient and parallel processing.
Siva Rajamanickam, a staff scientist and parallel computing expert at Sandia National Laboratories, highlights the suitability of MALA for scalable machine learning on HPC resources. The algorithm used in MALA can be easily distributed across different accelerators, maximizing speed and efficiency in electronic structure calculations.
The development of the MALA software stack has revolutionized electronic structure simulation by combining machine learning and physics-based approaches. This innovative method provides both accuracy and scalability, overcoming the limitations of existing techniques. With MALA, researchers can now simulate larger systems at unprecedented speeds, enabling advancements in various scientific fields. The future holds promising possibilities for using MALA to address societal challenges and drive further research and development.
Leave a Reply